
Is België klaar voor de nieuwe Europese regelgeving inzake de gezamenlijke gezondheidstechnologie evaluatie van innovatieve geneesmiddelen?
Binnen minder dan een jaar treedt de nieuwe Europese regelgeving voor evaluatie van gezondheidstechnologie (HTA = Health Technology Assessment) in werking, waarbij nieuwe oncologieproducten en geneesmiddelen voor geavanceerde therapie (ATMP's) als eerste in scope zijn vanaf 12 januari 2025. Deze verandering betekent dat de laatste sprint naar implementatie is begonnen.
Het dialoogvenster, zowel op Europees als nationaal niveau, is nu open, want tegen 2025 moet het nieuwe HTA proces klaar zijn.
Deze nieuwe EU HTA-procedure brengt significante veranderingen mee voor de beoordeling van nieuwe geneesmiddelen die niet meer uitsluitend nationaal maar via een gezamenlijk Europees proces zal lopen.
Een goede integratie van de EU HTA-regelgeving in België zal leiden tot betere en snellere toegang tot innovatieve technologieën, waarbij het RIZIV (Rijksinstituut voor Ziekte- en Invaliditeitsverzekering) een pioniersrol kan spelen op Europees niveau. De geïntegreerde aanpak tussen EU en nationale HTA zal efficiënt gebruik van middelen bevorderen, duplicatie vermijden en leiden tot meer helderheid en voorspelbaarheid in het beoordelingsproces, met vroege betrokkenheid van alle belanghebbenden voor een optimale afstemming.
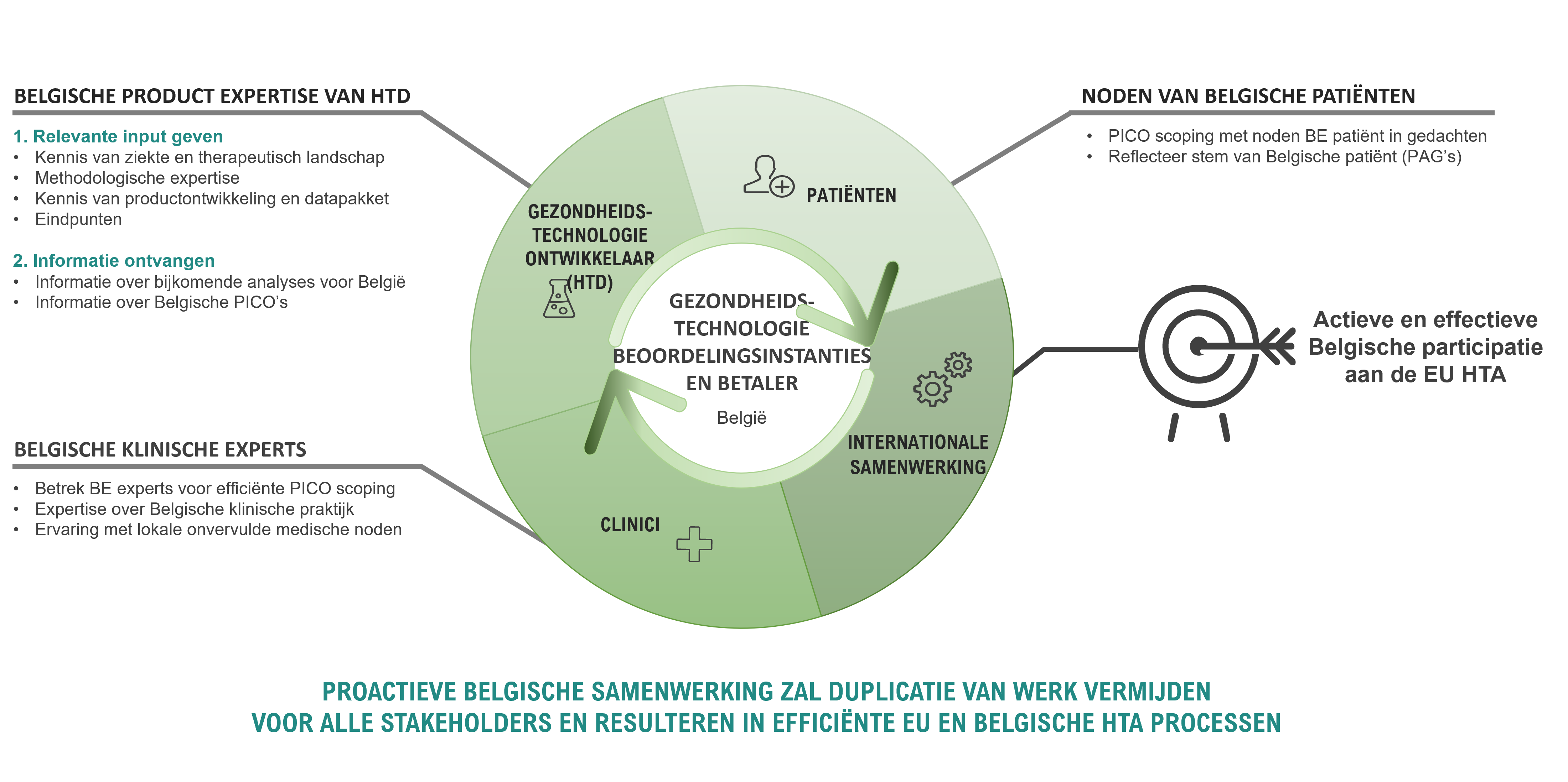
Via cocreatie met de verschillende belanghebbenden, waaronder het RIZIV, de Commissie Tegemoetkoming Geneesmiddelen (CTG) samen met farmaceutische bedrijven, patiënten en clinici, kunnen we deze implementatie tot een succes maken!
De toegang tot innovatieve technologieën voor Belgische patiënten is onze gezamenlijke topprioriteit.
Belangrijke veranderingen in de beoordeling van geneesmiddelen: Europese Health Technology Assessment (HTA) regelgeving vanaf 12 januari 2025 van toepassing
Health Technology Assessment (HTA) is een multidisciplinair proces dat de medische, sociale, economische en ethische aspecten van gezondheidstechnologieën evalueert. Het doel is om besluitvorming te ondersteunen.
De nieuwe EU HTA-regelgeving beoogt een einde te maken aan de inefficiëntie van het huidig gefragmenteerd systeem, waarbij meer dan 27 beoordelingen van dezelfde geneesmiddelen plaatsvinden op nationaal niveau, soms met identieke en soms met uiteenlopende vereisten en conclusies.
Met de oprichting van de EU HTA kunnen Europese lidstaten middelen, expertise en werklast delen, waardoor een gezamenlijke klinische analyse (Joint Clinical Assessment - JCA) en dit, desgevallend, in vergelijking met reeds beschikbare gezondheid technologieën. Deze analyse is gebaseerd op informatie die specifiek door de technologieontwikkelaar wordt verstrekt over zijn nieuw product. Het doel van de EU JCA is om de kwaliteit en tijdlijn van nationale evaluaties en toegangsbeslissingen te verbeteren.
De nieuwe EU HTA-regelgeving treedt effectief in werking op 12 januari 2025 en zal zich toespitsen op het klinisch deel van de HTA van geneesmiddelen en gezondheidstechnologieën. Deze evaluatie op Europees niveau zal parallel gebeuren met de Europese marktvergunningsprocedure. Het is de bedoeling dat ze op nationaal niveau gebruikt zal worden om zich verder uit te spreken over de waardebepaling van deze technologieën in een Belgische context.
Voor geneesmiddelen onder dit nieuwe proces, is het de bedoeling dat het RIZIV de (vergelijkende) klinische evaluatie niet opnieuw uitvoert.
Door deze wetgeving als een van de eerste administraties toe te passen, kan het RIZIV een voortrekkersrol spelen in Europa.
Om de JCA op nationaal niveau niet te dupliceren, en aanvullende analyses voor zeer uitzonderlijke gevallen te reserveren, is het essentieel de EU JCA structureel te integreren in de nationale terugbetalingsprocedure. Dit zorgt ervoor dat Europese en nationale processen optimaal op elkaar aansluiten en elkaar niet overlappen of vertragen.
Op het niveau van het RIZIV moeten voldoende middelen beschikbaar zijn om zowel op het Europese niveau als op het nationale niveau te verzekeren dat de verschillende fasen van de procedure binnen de voorziene tijd en op kwalitatieve wijze kunnen worden uitgevoerd.
Het ultieme doel is om de toegang tot innovatieve geneesmiddelen voor patiënten te verbeteren.
Wanneer zal deze uniforme Europese evaluatieprocedure van toepassing zijn voor alle innovatieve behandelingen?
De tijdlijn voor geneesmiddelen die moeten worden beoordeeld door een gezamenlijke klinische evaluatie is als volgt volgens de huidige officiële teksten:
- 2025: Nieuwe kankergeneesmiddelen en geneesmiddelen voor geavanceerde therapie (ATMP)
- 2028: Weesgeneesmiddelen
- 2030: Overige groep van geneesmiddelen [1]
Als we kijken naar de innovatiepijplijn van onze industrie, zien we dat er naar verwachting een breed scala aan geneesmiddelen en op de markt zal komen met een uniek potentieel om het leven van patiënten in de hele EU te verbeteren. Ontwikkelingsprogramma's richten zich steeds meer op kleinere, beter gedefinieerde doelpopulaties. Van de 41 marktvergunningen voor nieuwe werkzame stoffen vanwege het Europees Geneesmiddelenbureau in 2022 , waren er 18 voor nieuwe weesgeneesmiddelen of geneesmiddelen voor geavanceerde therapie (ATMP) [2].
Elke belanghebbende heeft een rol te spelen bij het succes van deze nieuwe procedure
Het is van essentieel belang om deze nieuwe procedure correct in te voeren om de toegang van patiënten tot innovatieve therapieën te versnellen. Vanaf het allereerste dossier op 12 januari 2025 moet het proces van hoge kwaliteit zijn.
Het betrekken van belanghebbenden in een gezamenlijke inspanning, met name bij het definiëren van methodologische en procedurele normen, van farmaceutische bedrijven tot patiënten en clinici, zal ervoor zorgen dat de beste expertise benut wordt. Door nu samen te werken kunnen we zorgen voor een gestroomlijnde en effectieve implementatie van het nieuwe systeem. Dit is cruciaal om ervoor te zorgen dat het proces soepel verloopt en dat de beloofde snellere toegang tot innovatieve therapieën voor patiënten wordt waargemaakt. Het is belangrijk te vermelden dat deze samenwerking niet alleen nodig is tijdens de huidige implementatiefase, maar ook gedurende het proces zelf, eenmaal geïmplementeerd. De inspanningen die vandaag worden geleverd, moeten worden benut voor een efficiënte voorbereiding van elk dossier vanaf 2025.
Op 30 januari 2024, heeft de Europese Commissie en de Heads of HTA Agencies Group (HAG), samen met nationale HTA-agentschappen van Nederland, Oostenrijk, België, Ierland en Luxemburg een bijeenkomst voor belanghebbenden georganiseerd die zich richtte op de nationale implementatie van de regelgeving van de Europese Unie betreffende de evaluatie van gezondheidstechnologie. Het evenement bracht de belangrijkste belanghebbenden samen, waaronder het RIZIV en vertegenwoordigers van nationale belanghebbenden zoals patiëntenverenigingen, gezondheidswerkers, wetenschappelijke verenigingen, vertegenwoordigers van de industrie, nationale besluitvormers en regionale gezondheidsautoriteiten.
Vroegtijdig overleg tussen alle belanghebbenden laat toe expertise, kennis en inzichten te bundelen en ervoor te zorgen dat de toegang van nieuwe effectieve, veilige en hoogwaardige geneesmiddelen voor patiënten in België in de nabije toekomst kan verbeteren.

Laten we samenwerken en in interactie treden, gezien onze bedrijven unieke ervaring hebben met diverse HTA-instanties en -methoden, waardoor we kansen voor harmonisatie kunnen benutten. Onafhankelijkheid van het HTA-proces is cruciaal, maar het is even belangrijk om de kennis van verschillende belanghebbenden te integreren in het nieuwe Europese systeem, waardoor we vanaf 2025 hoogwaardig gezamenlijk werk kunnen leveren.
[1] Details hierover worden verder verduidelijkt in de loop van 2024
[2] EMA Annual Report 2022

Schrijf je in op onze nieuwsbrief
Wil je op de hoogte blijven van het reilen en zeilen binnen de farma-industrie, schrijf je dan in op onze nieuwsbrief!