
La Belgique est-elle prête pour la nouvelle réglementation européenne sur l'évaluation des technologies de la santé pour les médicaments innovants ?
Dans moins d'un an, la nouvelle réglementation européenne sur l'évaluation des technologies de la santé (HTA – Health Technology Assessment) entrera en vigueur. Les nouveaux produits oncologiques et les médicaments de thérapie innovante (ATMP) entreront dans le champ d'application à partir du 12 janvier 2025. Ce changement signifie que la Belgique entre dans la dernière ligne droite pour sa mise en application.
La fenêtre de dialogue, tant au niveau européen que national, est maintenant ouverte, car le nouveau processus HTA doit être prêt pour 2025.
Cette nouvelle procédure européenne d’HTA apporte des changements significatifs pour l'évaluation des nouveaux médicaments, qui ne se fera plus exclusivement au niveau national, mais dans le cadre d'un processus européen commun.
L'intégration correcte des réglementations européennes en matière d'HTA en Belgique permettra d'améliorer et d'accélérer l'accès aux technologies innovantes, domaine dans lequel l’INAMI peut jouer un rôle de pionnier au niveau européen. L'approche intégrée entre l'UE et l'HTA nationale favorisera une utilisation efficace des ressources, évitera les doubles emplois et conduira à une plus grande clarté et prévisibilité dans le processus d'évaluation, avec l'implication précoce de toutes les parties prenantes pour un alignement optimal.
Un processus de co-création avec les différentes acteurs impliqués – l'INAMI, la Commission du remboursement des médicaments (CTG), les entreprises pharmaceutiques, les patients et les cliniciens – nous de réaliser cette mise en application avec succès !
L'accès des patients belges aux technologies innovantes est notre priorité commune.
Changements majeurs dans le processus d'évaluation des médicaments : le règlement européen sur l'évaluation des technologies de la santé (HTA) entrera en vigueur à partir du 12 janvier 2025
L'évaluation des technologies de la santé (HTA) est un processus multidisciplinaire qui évalue les aspects médicaux, sociaux, économiques et éthiques des technologies de la santé. Son objectif est d'aider à la prise de décision.
Le nouveau règlement européen sur l'évaluation des technologies de santé vise à mettre fin à l'inefficacité du système fragmenté actuel, dans lequel plus de 27 évaluations des mêmes médicaments ont lieu au niveau national, avec des exigences et des conclusions parfois identiques et parfois différentes.
Avec la création de l'HTA au sein de l’UE, les états membres peuvent partager leurs ressources, leur expertise et leur charge de travail, ce qui permet de réaliser une évaluation clinique conjointe (Joint Clinical Assessment - JCA). Cette analyse est basée sur des informations spécifiquement fournies par le développeur de technologie sur son nouveau produit. L'objectif de l'évaluation clinique conjointe de l'UE est d'améliorer la qualité et le calendrier des évaluations nationales et des décisions d'accès.
La nouvelle réglementation européenne l'évaluation des technologies de santé entrera en vigueur le 12 janvier 2025 et se concentrera sur la partie clinique de l'HTA des médicaments et des technologies de la santé (Joint Clinical Assessment - JCA), et, le si nécessaire, sur les technologies de santé déjà disponibles. Cette évaluation au niveau européen sera réalisée parallèlement à la procédure européenne d'autorisation de mise sur le marché. Il est prévu qu'elle soit utilisée au niveau national pour commenter l'évaluation de ces technologies dans le contexte belge.
Pour les médicaments soumis à ce nouveau processus, il est prévu que l'INAMI ne répète pas l'évaluation clinique (comparable). En étant l'une des premières administrations à appliquer cette législation, l'INAMI peut jouer un rôle de pionnier en Europe.
Afin de ne pas dupliquer l'analyse conjointe au niveau national et de réserver les analyses supplémentaires à des cas très exceptionnels, il est essentiel d'intégrer structurellement l'analyse conjointe de l'UE dans la procédure de remboursement nationale. Cela garantit que les processus européens et nationaux sont alignés de manière optimale et ne se chevauchent pas ou ne se retardent pas mutuellement.
Des ressources suffisantes doivent être disponibles au niveau de l'INAMI pour assurer que les différentes étapes de la procédure puissent être réalisées dans les délais prévus et de manière qualitative, tant au niveau européen qu'au niveau national.
L'objectif ultime est d'améliorer l'accès des patients aux médicaments innovants.
Quand cette procédure d'évaluation européenne uniforme s'appliquera-t-elle à tous les traitements innovants ?
Selon les textes officiels en vigueur, voici le calendrier des médicaments devant faire l'objet d'une évaluation clinique conjointe :
- 2025 : Nouveaux médicaments contre le cancer et produits médicinaux de thérapie avancée (ATMP)
- 2028 : Médicaments orphelins
- 2030 : Autre groupe de médicaments [1]
En examinant les innovations dans le pipeline de notre industrie, on peut s'attendre à ce qu'un large éventail de médicaments arrivent sur le marché avec un potentiel unique d'amélioration de la vie des patients dans l'ensemble de l'UE. Les programmes de développement se concentrent de plus en plus sur des populations cibles plus petites et mieux définies. Sur les 41 nouvelles substances actives dont l'Agence européenne des médicaments (EMA) a recommandé l'autorisation en 2022, 18 concernaient de nouveaux médicaments orphelins ou des médicaments de thérapie innovante (ATMP) [2].
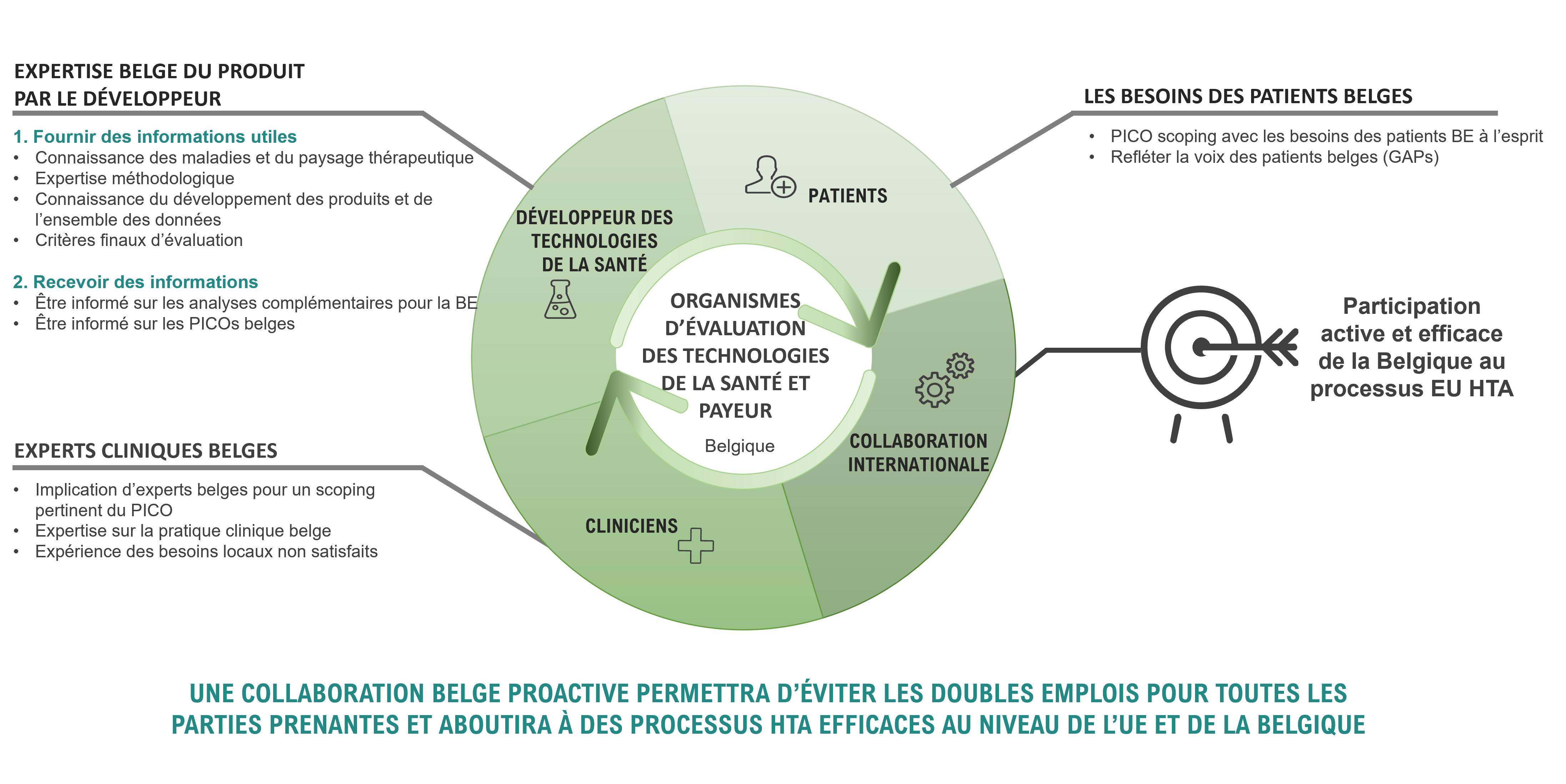
Chaque partie prenante a un rôle à jouer dans la réussite de cette nouvelle procédure
Il est essentiel que cette nouvelle procédure soit correctement implémentée pour accélérer l'accès des patients aux thérapies innovantes. Le processus doit être qualitatif dès l’examen du premier dossier, le 12 janvier 2025,
L'engagement des différents acteurs dans un effort commun, en particulier pour définir des normes méthodologiques et procédurales – des sociétés pharmaceutiques aux patients et aux cliniciens – permettra de garantir l’usage la meilleure expertise. En travaillant ensemble dès maintenant, nous pouvons garantir une mise en œuvre rationalisée et efficace du nouveau système, essentiel pour garantir le bon déroulement du processus et l'accès plus rapide aux thérapies innovantes promis aux patients. Il est important de mentionner que cette collaboration est nécessaire non seulement dans le cadre de la phase actuelle de mise en œuvre, mais aussi durant le processus-même, une fois d’application.
Les efforts déployés aujourd'hui seront utiles pour préparer efficacement chaque cas à partir de 2025.
Le 30 janvier 2024, la Commission européenne et le groupe des responsables d'agences d'HTA (HAG – Heads of HTA Agencies Group), ainsi que les agences nationales d'HTA des Pays-Bas, de l'Autriche, de la Belgique, de l'Irlande et du Luxembourg, ont organisé une réunion axée sur la mise en œuvre nationale des réglementations de l'Union européenne en matière d'évaluation des HTA. L'événement a rassemblé les principales parties prenantes, y compris l’INAMI et les représentants des parties prenantes nationales telles que les associations de patients, les professionnels de la santé, les associations scientifiques, les représentants de l'industrie, les décideurs nationaux et les autorités sanitaires régionales.
Des consultations précoces entre toutes les parties prenantes permettent de mettre en commun l'expertise, les connaissances et les idées et de garantir que l'accès des patients belges à de nouveaux médicaments efficaces, sûrs et de haute qualité puisse s'améliorer dans un avenir proche.

Travaillons ensemble et interagissons, car nos entreprises ont une expérience unique des différents organismes et méthodes d'HTA, ce qui nous permet d'exploiter les possibilités d'harmonisation. L'indépendance du processus HTA est cruciale, mais il est tout aussi important d'intégrer les connaissances des différentes parties prenantes dans le nouveau système européen, ce qui nous permettra de réaliser un travail commun de grande qualité à partir de 2025.
[1] Les détails seront précisés dans le courant de l'année 2024
[2] EMA Annual Report 2022

Intéressé(e) à recevoir les nouvelles de pharma.be
Etre au courant de ce qui vit au sein de l'industrie pharmaceutique ? Inscrivez-vous à notre lettre d'information.